


MedFriendly®
















Adrenoleukodystrophy
Adrenoleukodystrophy (abbreviated as ALD) is a rare
disorder that is characterized by a buildup of saturated,
very long chain fatty acids (abbreviated VLCFA) in the
tissues of the body. Saturated means to be filled up
with something. In the case of fats, saturated refers to
how much hydrogen (a type of element) is contained in
it. Saturated fats are filled with hydrogen. Fatty acids
are types of chemicals in the body that are necessary
for many bodily functions. Very long chain fatty acids
are called "very long" because they have greater
amounts of carbon in them. Carbon is a type of
element that is essential to normal bodily functioning.
FEATURED MOVIE ON
ADRENOLEUKODYSTROPHY: Lorenzo's Oil
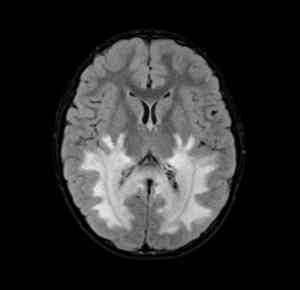
White matter damage in the
brain (white areas) caused by
adrenoleukodystrophy.
In ALD, very long chain fatty acids are found in especially high numbers in the brain,
blood, and the adrenal cortex (the outer part of the adrenal glands). The adrenal glands
are a pair of glands that play an important role in metabolism and help the body respond
to physical and emotional stress by releasing certain hormones. A gland is an organ in
the body made of special cells that form and release materials such as fluid. Metabolism
is the chemical actions in cells that release energy from nutrients or use energy to
create other substances. Hormones are natural chemicals produced by the body and
released into the blood that have a specific effect on tissues in the body. In ALD, the
buildup of very long chain fatty acids cause the adrenal glands to waste away and
decrease in size.
"Where Medical Information is Easy to Understand"™
Since the adrenal glands waste away, metabolism becomes
impaired. This is why ALD is often called a metabolic disease.
In ALD, the buildup of very long chain fatty acids also causes a
widespread loss of myelin in the brain. Myelin is a fatty nerve
covering that helps nerve impulses travel quickly. This function
will become impaired when myelin is destroyed. The loss of
myelin causes inflammation, which destroys nerve cells.
WHAT ARE THE SIGNS AND SYMPTOMS OF
ADRENOLEUKODYSTROPHY?

The general signs and symptoms vary depending on the type of ALD (see the next section). This section
describes some more common signs of ALD. Some signs of ALD include progressive and severe
intellectual impairment, impaired language functioning, eventual blindness, muscle spasms, quadriplegia,
and apraxia. Quadriplegia is a loss of the ability to move and/or feel both arms, both legs, and the parts
of the body below the area of injury or damage to the spinal cord, which is what causes the condition to
occur.
Apraxia is an impairment in the ability to perform voluntary skilled movements or purposeful acts in a
person with intact motor abilities. The person may have difficulty performing the movements necessary to
dress or construct things. Apraxia is due to damage to the parietal lobes of the brain. The parietal lobes
are located in top, upper back part of the brain. In addition to apraxia, some patients with ALD have
spastic paralysis. Spastic paralysis is a loss of muscle function in addition to contractions or shortening
of the muscles.
An insufficient production of hormones by the adrenal glands is another sign of ALD. This can lead to
nausea, vomiting, weakness, weight loss, a craving for salt, and postural hypotension. Postural
hypotension is low blood pressure in which symptoms (such as lightheadedness and dizziness) become
present upon standing. Insufficient adrenal glad functioning can cause a failure to thrive, which is an
abnormal slowing of growth and development of the infant.
Insufficient adrenal gland functioning causes a mildly increased coloring of the skin, especially in the lips,
nipples, inner cheek, scar tissue, joints, and the mucous membrane lining inside the mouth. A joint is a
place where two bones contact each other. A mucous membrane is one of four major types of thin
sheets of tissue that line or cover various parts of the body.
It should be mentioned that about 33% of patients with ALD do not have neurological signs present,
meaning that they do not present with problems (such as loss of vision and paralysis) due to brain and/or
spinal cord damage. For details on the most common signs and symptoms in specific types of ALD, see
the next section.
ARE THERE DIFFERENT TYPES OF ADRENOLEUKODYSTROPHY?
Yes. These are several different forms of ALD, some of which are described below:
CLASSIC CHILDHOOD FORM: The classic childhood form of ALD occurs is the most severe form of
ALD and occurs only in males (the reason for this is described in the next section). It is usually seen
between the ages of 4 and 10 (most commonly between 4 and 8). The average age that this form of ALD
occurs is age 7. Although this form of ALD very rarely occurs earlier than age 3, it has been known to
occur as early as age 2. This form of ALD very rarely occurs over age 15. About 35% of all cases of
ALD are the classic childhood form.
At first, the child with this form of ALD develops normally, usually up to 3 and 4 years of age. The most
common problems usually begin with behavioral changes such as hyperactivity, difficulty paying
attention, aggression, poor academic performance, worsening handwriting, difficulty reading, poor
comprehension of written information, and abnormal withdrawal. Because of these behavioral problems,
these children are often misdiagnosed with Attention-Deficit/Hyperactivity Disorder (ADHD), a mental
disorder characterized by overactivity and/or difficulties paying attention. Although the behavioral
problems may respond to medications that stimulate the child, which are used to treat ADHD, this does
not mean that the child has ADHD.
After the behavioral changes, more serious difficulties occur such as loss of hearing and loss of vision.
The hearing loss may become noticeable by the child having difficulty discriminating sounds in a noisy
room or difficulty understanding speech on the telephone (even though perception of sounds is normal).
The hearing loss can eventually progress to deafness. The visual loss can be characterized by the child
not being able to see visual information on the right or left side. The person may not be able to see
clearly, may have double vision, or may experience strabismus. Strabismus is a condition in which there
is an abnormal deviation of one eye in relation to the other.
Other signs of this form of ALD include difficulty swallowing (known as dysphagia), learning impairments,
poor memory, fatigue, increased coloring of the skin, occasional vomiting, poor coordination, difficulty
walking, clumsiness, apraxia (see last section), seizures, and dysarthria.
Seizures are involuntary muscle movements and/or decreased awareness of the environment due to
overexcitement of nerve cells in the brain. About 33% of patients with this form of ALD develop seizures
and it may be the first sign of ALD in some patients with this form of the disease. Dysarthria is a difficulty
in speech articulation that results from an impaired ability to control the muscles involved in speech.
Progressive and severe intellectual impairment can also occur in the childhood form of ALD. About 90%
of children with this form of ALD have an insufficient production of hormones by the adrenal glands by the
time signs of the disease are first noticed.
Some children with this form of ALD develop impaired spatial abilities, astereognosis, and graphesthesia.
Asteroegnosis is an inability to recognize objects by touching them even though the sense of touch and
proprioception is intact. Proprioception is the sense of being aware of the position and movements of the
body. After these problems occur, signs related to adrenal gland dysfunction (discussed in the prior
section) occur.
The signs of ALD can progress rapidly although the rate of progression is variable in different patients.
Over time, the paralysis and increased muscle tension increases. Visual and hearing loss worsens and
there is loss of the ability to swallow or speak. Intellectual impairment also gets worse.
This form of ALD can lead to a persistent vegetative state within 6 months to 2 years after symptoms
begin. A persistent vegetative state is a type of indefinite deep coma. A coma is a state of deep
unconsciousness in which there are no voluntary movements, no responses to pain, and no verbal
speech. The persistent vegetative state can last for more than 10 years. When a person is in a
persistent vegetative state he/she is totally disabled. Death can occur anytime after symptoms begin, but
usually occurs in a time frame of 1 to 10 years.
ADOLESCENT (CEREBRAL) ALD: This form of ALD begins between age 10 and 21. The signs are the
same as childhood ALD, but they progress at a slower rate.
ADULT-ONSET FORM (ADRENOMYELONEUROPATHY): This is a milder form of ALD and can occur
between the ages of 21 and 35 (most commonly in the late 20s). This form of ALD progresses more
slowly than the childhood form. However, about half of the men with this form of ALD experience impaired
brain functioning. Signs and symptoms of adult-onset ALD usually begin with stiffness and weakness in
the legs that gets worse over time. Many patients with this form of ALD develop progressive (worsening)
spastic paraparesis. Spastic paraparesis is weakness in the legs in addition to contractions or
shortening of the leg muscles.
Other signs and symptoms of this type of ALD include ataxia (impairment in coordinating movement),
difficulty walking, sexual dysfunction, and sensory loss. About half of patients with this form of ALD show
signs of brain damage on brain scans or through a physical examination. About 33% of people with this
form of ALD have myelin loss. About 10 to 15% of females with ALD have signs consistent with damage
to the brain and/or spinal cord. In 10 to 20% of patients with this form of ALD, the brain damage becomes
severe, which can lead to severe behavioral and cognitive (thinking) problems. The brain damage can
result in total disability and death.
In males with adrenomyeloneuropathy, there is a loss of myelin in the nerve tracts in the spinal cord. The
loss of myelin in the spinal cord causes difficulties walking and also leads to difficulties controlling bladder
and bowel movements. The difficulty controlling the bowels is due to a loss of sphincter control. A
sphincter is a muscle that forms a circle around a tube or natural opening in the body.
About 70% of adults with this form of ALD have an insufficient production of hormones by the adrenal
glands. This form of ALD usually progresses slowly over a 5 to 15 year period, but can go on progressing
for decades.
NEONATAL ADRENOLEUKODYSTROPHY: This form of ALD is often abbreviated as NALD. NALD is
present from birth and can occur in males and females. NALD usually progresses quickly and may result
in mental retardation, facial abnormalities, decreased tension in the muscles, impairment of the adrenal
glands, seizures, enlarged liver, and degeneration of the retina.
The liver is the largest organ in the body and is responsible for filtering (removing) harmful chemical
substances, producing important chemicals for the body, and other important functions. The retina is an
area at the back of the eye that is sensitive to light.
ADDISON'S ONLY FORM: In this form of ALD, only the adrenal glands seem to be involved and the brain
and spinal cord seem to be spared. However, later on in this form of ALD, the signs appear similar to the
adult-onset form of the disease (see above). In this form of ALD, the dysfunction of the adrenal glands is
usually apparent by age 7 and a half but can occur anytime between age 2 and adulthood. Females with
this form of ALD normally have a milder case than males. Common signs for this type of ALD include
unexplained vomiting, weakness, or coma. Increased skin coloring may or may not be present.
FEMALE FORM: Although not a specific type of ALD, it is worth pointing out again that women with ALD
generally have mild signs and symptoms of the disease. In women, signs and symptoms generally include
increased muscle tension, problems urinating, ataxia, mild to moderate spastic paraparesis (beginning
middle age or later), and mild peripheral neuropathy. Peripheral neuropathy is damage to nerve fibers
outside of the brain or spine. Many of the signs appear similar to the adult onset-form in males later on in
the female's life. Functioning of the adrenal glands is almost always normal in this form of ALD.
WHAT CAUSES ADRENOLEUKODYSTROPHY?
This section is broken into two parts: 1) The genetic defect and 2) The effects of the genetic defect.
THE GENETIC DEFECT: ALD is a genetic disorder, meaning that there is a defect in one of the patient's
genes. Genes are units of material contained in a person's cells that contain coded instructions as for
how certain bodily characteristics (such as eye color) will develop. All of a person's genes come from
his/her parents.
Genes can either be dominant or recessive. A gene that masks the effect of another gene is called a
dominant gene. The gene whose expression is masked is known as a recessive gene. There are many
different forms of genes. Each form is known as an allele. Some alleles are normal whereas others may
be abnormal. Abnormal alleles can cause diseases, such as ALD. To fully understand how genes can
cause ALD, a more detailed description of genes is required.
Genes are contained in structures called chromosomes. Each person has 23 pairs of chromosomes,
meaning that there are 46 chromosomes in total. One of each pair of chromosomes is inherited from the
mother and one of each pair is inherited from the father. The first 22 pairs of chromosomes (known as
autosomes) are not involved in determining sex. The 23rd pair of chromosomes, however, is involved in
determining sex.
The 23rd pair of chromosomes consist of X and/or Y chromosomes. An X chromosome is shaped like an
X, whereas a Y chromosome is shaped like a Y. If a person has two X chromosomes, the person will
develop as a female. If a person has an X and a Y chromosome, the person will develop as a male.
The X chromosome is much larger than the Y chromosome. The X chromosome has a few thousand
genes whereas the Y chromosome has only a few genes. For this reason, most of the genes on the X
chromosome have no counterpart on the Y chromosome. The genes of the Y chromosome are not
capable of masking the expression of genes from the X chromosome. In females, however, the genes of
one X chromosome are capable of masking the effects of the genes on the other X chromosome.
If there is a disease causing gene (such as the gene for ALD) on the X chromosome, a female has the
extra protection of the other X chromosome to mask its effect, whereas the male does not have this
protection. Although the expression of recessive genes is normally masked by its dominant counterpart, if
no such counterpart is present, even a recessive gene passed on from the mother will express itself.
In females, one X chromosome is inherited from the mother and one from the father. In males, the X
chromosome is inherited from the mother and the Y chromosome is inherited from the father. Thus, in
males who have diseases due to abnormal genes on the X chromosome, the disease has been passed
on from mother to son. The reason why the mother does not have the disease is because she has a
dominant gene on the second X chromosome that protects her from the effect of the recessive gene that
causes ALD.
About 93% of ALD cases are due to a faulty gene being passed on from one parent to the child. About
7% of cases are due to a fault in the gene occurring spontaneously in the child. Mothers who carry the
defective gene that cause ALD have a 50% chance of passing it on to their child. The mother's son will
always get the disease. The mother's daughter will become a carrier of the defective gene, but likely
won't develop a serious form of ALD. All daughters of a father who carries the gene for ALD will carry this
gene as well. None of the sons of a father who carries the gene for ALD will carry this gene.
The gene that causes ALD was discovered in 1993. It is known as the ABCD1 gene. There are over 200
ways in which the ABCD1 gene can be abnormal. The ABCD1 gene is located on the X chromosome.
The X chromosome can be divided into different parts such as part a (known as Xa) or part b (known as
Xb), etc. The gene that causes ALD is located on part q (the end part) of the X chromosome (abbreviated
Xq).
Genes contain substances known as nucleotides, which are building blocks that contain the code for how
certain proteins are produced. The nucleotides are assembled in a specific order based on the type of
protein that needs to be produced. It is much like how the letters of a word (such as "b-o-o-k-s") need to
be put in the correct order so that the word ("books") is spelled correctly. When the nucleotides are not
assembled correctly (like spelling the word "book" as "bokos") or if some of the nucleotides are deleted
(like spelling the work "book" as "bks") this results in the production of abnormal proteins. The production
of abnormal proteins can have devastating consequences to the body, one of which is ALD. The next
section will discuss the protein that is abnormal in ALD.
Genes can express themselves at different points in time. In the neonatal form of ALD, the responsible
gene expresses itself at birth. In the classic childhood form, the responsible gene expresses itself in
childhood. In the adult-onset form (adrenomyeloneuropathy), the responsible gene does not express itself
until adulthood.
THE EFFECTS OF THE GENETIC DEFECT: To understand the effects of the genetic defect that causes
ALD, it is first necessary to understand the role of peroxisomes. Peroxisomes are structures present in
almost all cells, and help to rid the body of poisonous substances. There are a high number of
peroxisomes present is specialized cells that produce myelin.
Peroxisomes contain enzymes that break down very long chain fatty acids. Enzymes have bindings sites
for the chemicals that they interact with. Picture a binding site on an enzyme as a keyhole and the
chemical that the enzyme interacts with as a key. Just like only a certain type of key will fit in a keyhole,
only a certain type of chemical will fit in a particular enzyme.
The enzyme that breaks down fatty acids has a binding site on it for fatty acids and a binding site on it for
another chemical, known as coenzyme A (abbreviated CoA). When these two chemicals come together
in the enzyme, a new product is formed, known as fatty acyl-CoA. The breakdown of very long chain fatty
acids is dependent on this first step. Without the enzyme present, there is almost no chance that CoA
and fatty acids would ever come together.
Only the enzyme found in peroxisomes can break down very long chain fatty acids. This enzyme is known
as VLCS (very long chain fatty acyl CoA systhesase). The enzymes that break down shorter chains of
fatty acids function normally. However, the VCLS enzyme does not function normally in patients with
ALD. Because of this, researchers suspected that the gene that causes ALD would direct the body to
make this enzyme. To their surprise this was not the case.
In 1993, researchers (Drs. Patrick Aubourg and Jean-Louis Mandel) in Paris discovered that the genes
responsible for ALD directs the body to make a protein that works differently than an enzyme. This
protein is known as ALDP (adrenoleukodystophy protein) and it is a type of transporter protein.
Transporter proteins help carry substances in and out of cells or in an out of structures that are inside of
cells.
ALDP is located in the membrane (outer covering) of peroxisomes. It is believed that ALDP normally
helps to transport very long chain fatty acids into the peroxisome where it can be broken down. In ALD,
the defective gene produces a defective version of ALDP. This defective version is not able to transport
very long chain acids into the peroxisomes to be broken down. As a result, the very long chain fatty acids
build up and cause damage in the body.
Specifically, very long chain fatty acids build up in the rough endoplastic reticulum of tissues in the body,
causing a toxic effect. Endoplasttic reticulum is a complex system of folded, flat sacs that provide a large
area for fluid to be stored and for reactions to occur. Endoplastic reticulum with ribosomes in them is
known as rough endoplastic reticulum. Ribosomes are small, round particles that build up proteins.
In the adrenal cortex, the increased levels of very long chain fatty acids results in a decreased capacity
to convert cholesterol into the hormones normally produced by the adrenal glands. Cholesterol is a waxy,
fatty substance found only in animal tissues.
As was mentioned previously, the buildup of fatty acids appears to be what causes the destruction of
myelin. How this happens is not completely understood. It is also not completely understood how ALDP
affects the enzyme that breaks down fatty acids.
One possible mechanism by which very long chain fatty acids get to interact with myelin can be explained
by the structure of these fatty acids. Specifically, very long chain fatty acids are saturated, meaning that
they are filled with hydrogen. Saturated fatty acids are straight in structure. Their straight structure allows
them to interact with myelin better than fatty acids that are not straight. The very long chain fatty acids
may build up in the myelin, causing the body's defense system to respond. It is possible that inflammation
may result, which then destroys the myelin.
HOW IS ADRENOLEUKODYSTROPHY DIAGNOSED?
Since very long chain fatty acids accumulate in the blood in patients with ALD, this can be detected with
a blood test. The findings of the blood test are combined with the history of the presenting problems and
the specific signs that the patient presents with to make a diagnosis. The blood test reveals the
presence of very high levels of very long chain fatty acids. This blood test is extremely specialized, and
as a result, only a few laboratories in the world can do it. However, it is the most important laboratory
test used to diagnose ALD.
There are also very high levels of very long chain fatty acids in samples of fibroblasts that are grown in a
culture (a special laboratory container). Fibroblasts are flat, long cells in connective tissues that give rise
to other types of cells. Connective tissues are tissues that connect other tissues and body parts.
Very high levels of very long chain fatty acids are found in about 99.9% of males with ALD, regardless of
age. About 85% of females have very high levels of very long chain fatty acids in the blood or in skin
fibroblasts. In pregnant mothers undergoing amniocentesis, high amounts of very long chain fatty acids
may be found. Amniocentesis is a procedure in which a small amount of fluid (known as amniotic fluid) is
withdrawn from the sac that surrounds the unborn child in the mother's uterus. This fluid can then be
tested to detect abnormalities in the unborn child. The uterus is a hollow organ in a female's body where
the egg is implanted and the baby develops.
After amniocentesis is performed, DNA (deoxyribonucleic acid) can be taken out of the cells from the
unborn child to identify if the gene that causes ALD is present. DNA is a chain of many connected genes.
If this type of genetic testing is not possible, amniocytes or cells from the chorionic villus can be taken
during amniocentesis and tested for the level of very long chain fatty acids. See the next paragraph for a
description of amniocytes and chorionic villus cells.
The results from the amniocentesis in mothers who are pregnant with a baby that will have neonatal ALD
typically reveals very long chain fatty acids in these cells. Amniocytes are cells in the amniotic fluid.
Before babies are born, when they are in the earliest stages of development, villi (tiny, fingerlike
structures) are found on the surface of the outermost membrane of the organism. This outermost layer is
known as the chorion. These type of villi are known as chorionic villi. Chorionic villi help form the
placenta, an organ in a female that nourishes a baby during pregnancy. The chorionic villi cells are
normally sampled while the mother is 10 to 12 weeks pregnant.
There have been occasions in which the testing of very long chain fatty acids in amniocytes and
chorionic villus cells have been normal when the child with neonatal ALD is born. This may have been
related to technical factors, however.
There are abnormal levels of the number of C26 fatty acids compared to C22 fatty acids and abnormal
levels of C24 fatty acids compared to C22 fatty acids. The initial "C" stands for the element, carbon. The
number after the C (such as 26) is the number of carbon atoms in the chain of fatty acids. Atoms are the
smallest part of an element that can exist alone or in combination with something else.
The very long chain acids C26 (also known as hexacosanoic), C25 (also known as pentacosanoic), and
C24 (also known as tetracosanoic) are present in the highest amounts in patients with ALD. The body
normally shortens fatty acids in the peroxisomes (see last section) by taking away two of the carbon
atoms.
The insufficient functioning of the adrenal glands can cause other problems that show up on laboratory
tests, such as decreased levels of salt. The decreased levels of salt in the body is why many patients
with ALD have a salt craving. Other problems caused by insufficient functioning of the adrenal glands
include mild acidosis (too much acid in the blood) and decreased levels of potassium. Potassium is a
metallic element that is necessary for animals to live. It plays an important role in helping adjust the
amount of water in the body, move muscles, and conduct nerve impulses.
Insufficient functioning of the adrenal glands causes a low level of cortisol to be present in the blood.
Cortisol (also known as hydrocortisone) is a type of hormone produced by the adrenal glands that helps
reduce inflammation. ALD patients have high levels of ACTH (adrenocorticotropic hormone in the blood).
ACTH is a type of hormone released in the brain that directs the adrenal glands to produce certain
hormones such as cortisol. ACTH is present is a high level because it is trying to produce more cortisol.
85% of ALD patients show an impaired response of cortisol in the presence of ACTH. Normally, levels of
cortisol increase in response to ACTH. The increased ACTH level is what causes increased skin coloring
in some patients.
WHAT DO BRAIN SCANS OF PATIENTS WITH ADRENOLEUKODYSTROPHY SHOW?
Even early on in the disease, the brains of patients with ALD may have significant abnormalities even if
the signs of the disease are mild. The types of tests used to get pictures of the inside of the brain are
known as CT scans and MRI scans. CT (computerized tomography) scanning is an advanced imaging
technique that uses x-rays and computer technology to produce more clear and detailed pictures than a
traditional x-ray. MRI (Magnetic Resonance Imaging) scans produce extremely detailed pictures of the
inside of the body by using very powerful magnets and computer technology.
Myelin loss is seen in about 85% of brain scans of patients with ALD, with the loss of myelin
approximately equal on both sides of the brain. Damage occurs to periventricular white matter.
Periventricular white matter refers to white matter that is immediately to the side of the two lateral (side)
ventricles of the brain. The lateral ventricles are two curved openings (shaped like a horseshoe) located
deep within the top section of the brain.
White matter is a group of white nerve fibers that conduct nerve impulses quickly. White matter is
important for muscle movements. The white matter that is affected in ALD may become hardened as
calcium deposits form in it. Calcium is a natural element that is very important in bone formation.
The periventricular white matter that is affected in ALD is located in the parietal and occipital lobes of
the brain. The parietal lobes are located in the top, upper back part of the brain. The occipital lobes are
located in the back part of the brain. About 12% of patients have damage to the frontal lobes (the front
section of the brain).
In some people with ALD, the splenium of the corpus callosum wears away. The corpus callosum is a
large band of nerve fibers in the brain that help the two sides of the brain communicate with each other.
The splenium is the thickened part in the back of the corpus callosum. As was mentioned earlier, nerve
pathways from the brain to the spine become damaged.
Another part of the brain that gets damaged in ALD is the thalamus. The thalamus is a pair of large oval
structures that sends out messages regarding sensation. Other parts of the brain affected in ALD are
the lateral geniculate bodies and the medial geniculate bodies. These structures are located close to the
thalamus and play an important role in vision and hearing, respectively.
Lymphocytes can also cross the blood brain barrier, a process known as lymphocyte infiltration. A
lymphocyte is a type of white blood cell present in the blood. White blood cells help protect the body
against diseases and fight infections. The blood brain barrier is a protective barrier produced by specific
types of cells in the brain that prevents many substances from entering the brain. Lymphocytes enter
the brain because ALD causes parts of the blood brain barrier to break down. The damage to the brain
continues to get worse over time and parts of the brain continue to wear away.
HOW IS ADRENOLEUKODYSTROPHY TREATED?
Unfortunately, there is no cure for ALD. However, in patients who are in early stages of the childhood
form of ALD and have mild/moderate brain involvement (and brain-related signs), the disease can be
treated with a bone marrow transplant. Bone marrow is a type of tissue that fills the inside of bones. The
reason for replacing defective bone marrow with normal bone marrow is that normal bone marrow
contains the protein that is deficient in ALD. Please see the section on causes of ALD to understand the
role of this protein.
The results of a bone marrow transplant is most favorable in patients with a Performance IQ over 80.
Performance IQ refers to one's ability to perform mostly novel, nonverbal tasks, as opposed to verbal
tasks. A score of 80 is in the low end of the low average range. The problems with bone marrow
transplants with ALD patients is that most people die within the first year of treatment.
Bone marrow transplants are not used for patients with severe brain damage or patients who have
evidence of brain damage on brain scans but no evidence of brain related signs (such a vision or
hearing loss). Bone marrow transplants are also not used in patients who do not have any evidence of
brain involvement or in patients with the adult-onset form of the disease who have myelin loss in the
brain. Bone marrow transplants have poor results in people with severe cases of ALD and mild results in
those with less than severe cases.
Physical therapy, special education, and psychotherapy are also important forms of treatment for ALD.
For patients with difficulty swallowing, soft and mushy foods are used. Some doctors have attempted to
reduce inflammation and improve metabolism by using statin drugs. Statin drugs block an enzyme the
body needs to make cholesterol. An enzyme is a type of protein that helps produce chemical reactions
in the body. Cholesterol is a waxy, fatty substance found only in animal tissues.
To treat the insufficient functioning of the adrenal glands, doctors treat ALD patients with hormones.
This can be a lifesaving treatment. The goal is to provide the patient with the hormones that are not
being provided by the adrenal glands. The hormones are usually taken by mouth in pill form. These
hormones are known as cortisone acetate and fludrocortisone. This type of treatment has no effect on
problems related to brain and spinal cord damage. The functioning of the adrenal glands is monitored
regularly by the doctor.
Seizures are treated with anticonvulsants. Anticonvulsants are medications used to prevent seizures.
The muscle relaxant drug, Baclofen, is used to treat painful muscle spasms. A sleeping pill called chloral
hydrate is sometimes given to help ALD patients sleep because many patients with this condition have
disrupted sleep.
There is a dietary treatment of ALD which was popularized in the excellent movie, Lorenzo's Oil. The
movie is about a parent's incredible fight to save the life of their child (Lorenzo Odone) who was
diagnosed with ALD.
The goal of the dietary treatment is to reduce the amount of very long chain fatty acids in the body. To
do this, the diet has to be very restrictive because many foods contain very long chain fatty acids, such
as nuts, milk, grains, fruits, vegetable skins, spinach, and traditional fattening foods (such as meat and
fried foods).
People who use the dietary treatment use a type of oil known as Lorenzo's Oil. Lorenzo's Oil is a
combination of two different oils known as glycerol trioleate oil and glycerol trierucate oil. There are four
parts of glycerol trierucate oil to each part of glycerol trioleate oil. Glycerol trioleate oil is found naturally
in rapeseed oil. Rapeseed is a type of mustard crop. Glycerol trierucate oil is made up of 90% oleic oil.
Oleic oil is found in sunflower seeds, olive oil, and corn oil. Oleic oil is an unsaturated fatty acid,
meaning that it is not filled with hydrogen. Lorenzo's Oil helps to quickly reduce a very long chain of fatty
acids in the blood. Specifically, there is about a 50% decrease in very long chain fatty acids by 4
months. Levels of the very long chain fatty acid, C26, may reach normal levels by 4 weeks. However,
Lorenzo's Oil appears to have little effect in stopping the progression of brain-related problems in the
disease.
HOW DOES LORENZO'S OIL REDUCE VERY LONG CHAIN FATTY ACIDS?
To answer this question, it is necessary to understand that there are two types of very long chain fatty
acids. There are saturated very long chain fatty acids and unsaturated very long chain fatty acids.
Saturated very long chain fatty acids are filled with hydrogen and are harmful to the body in excess.
Unsaturated very long chain fatty acids are not filled with hydrogen and are not harmful to the body.
The same enzyme makes both saturated and unsaturated very long chain fatty acids. If oleic oil is
available in the body, this enzyme will use it to make unsaturated very long chain fatty acids. If oleic
acid is not available in the body, this enzyme will use saturated fatty acids to make saturated very long
chain fatty acids. Since Lorenzo's Oil is mostly made of oleic oil, the production of unsaturated very long
chain fatty acids will increase and the production of the harmful saturated very long chain fatty acids will
decrease.
To boost the effects of oleic acid's ability to reduce the production of saturated very long chain fatty
acids, another oil was needed. This is why glycerol trioleate (GTO) was used. The enzyme that normally
makes saturated and unsaturated very long chain fatty acids will use glycerol trierucate (GTE) oil to
make unsaturated very long chain fatty acids instead of the saturated ones.
With both oleic oil and GTE oil present, saturated fatty acids are not able to bind to the enzyme that
makes saturated very long chain fatty acids. Lorenzo's Oil has the potential to reduce the number of
very long chain fatty acids to normal levels.
WHAT IS THE PROGNOSIS FOR PEOPLE WITH ADRENOLEUKODYSTROPHY?
The prognosis for males with ALD is unpredictably variable and each case is different. In general,
however, the prognosis for people with ALD is poor, except for patients that respond well to a bone
marrow transplant (see treatment section). If the disorder is present since birth, death usually occurs by
age 1 to age 5. Otherwise, death usually occurs within 1 to 10 years after the onset of symptoms. There
are exceptions, however, and most men with the adult-onset form of ALD maintain successful personal
and professional lives. Females with ALD generally have a mild form and are not seriously affected.
HOW MANY PEOPLE HAVE ADRENOLEUKODYSTROPHY?
Approximately 1 out of every 20,000 to 50,0000 people have ALD. The disease appears to be present
in equal amounts in all ethnic groups.
IS IT POSSIBLE FOR MOTHERS TO KNOW IF THEY CARRY THE GENE FOR ALD BEFORE
GETTING PREGNANT?
Yes. The first step is to have a blood test for very long chain fatty acids. If the very long chain fatty acid
level is abnormally high, the mother is a carrier of the defective gene that causes ALD. Since 15% of
female carriers have normal levels of very long chain fatty acids, genetic testing will need to be used to
determine if the mothers carries the gene that causes ALD. Your doctor should be able to direct you to
a genetic testing laboratory in your area. It is important to note that if a mother has a child with a mild
form of ALD, this does not mean that another child will not develop a severe form of this disease.
IF A MOTHER KNOWS THAT SHE CARRIES THE GENE FOR ALD AND HAS A SON, SHOULD HE
BE TESTED?
There is no clear answer to this question. What to do depends on each specific case and consultations
with your doctor. It is appropriate for parents to consider identifying a child that is at risk for ALD. The
testing would measure the level of very long chain fatty acids in the blood.
Benefits of identifying a child who will develop ALD is that insufficient functioning of the adrenal glands
can be treated early. This can potentially help avoid life-threatening complications related to insufficient
adrenal gland functioning. Early diagnosis can save the parents the time and money involved in trying to
determine the cause of the brain-related symptoms that develop.
It is important to keep in mind that only 35% of males with the gene for ALD will develop this disease
during childhood. This means that males are more likely to develop ALD after childhood. If the child is
diagnosed with ALD through such testing, but has not yet developed signs of ALD, the parents need to
consider whether to tell the child. These are difficult issues to make a decision about since no definitive
treatment exists.
The child will likely experience stress in knowing that he/she will develop a disease at an unknown time
in which there is no cure. If others know the diagnosis, the child may be stigmatized by friends, family,
and people in other social settings. Future academic and employment possibilities can also be affected.
As was mentioned earlier, amniocentesis can be performed in pregnant mothers to try and detect if the
baby will have ALD. Please click the link in the last sentence to read more about this procedure as it
relates to ALD diagnosis.
WHY AREN'T ALL NEWBORNS TESTED FOR HIGH LEVELS OF VERY LONG CHAIN FATTY
ACIDS?
When babies are born, it is normal to have high levels of very long chain fatty acids. Thus, finding high
levels of very long chain fatty acids in newborns would not mean that they had ALD. Also, as was
mentioned earlier, there are only a few laboratories in the world that are able to perform the specialized
tests needed to identify high levels of very long chain fatty acids.
HOW IS ADRENOLEUKODYSTROPHY CLASSIFIED?
ALD is classified as one of the leukodystrophies, meaning that it is a disorder that causes damage to
myelin in the brain.
WHERE CAN I GET MORE INFORMATION ON ADRENOLEUKODYSTROPHY?
For more information on adrenoleukodystophy, contact the United Leukodystrophy Foundation. You can
also contact the Myelin Project, a foundation established by the parents of a child with
adrenoleukodystrophy. These parents helped research a treatment for this condition discussed above,
which is documented in the excellent movie, Lorenzo's Oil. Contacting the Myelin Project can lead you to
find help in your area of the country.
HOW IS ADRENOLEUKODYSTROPHY ABBREVIATED?
Adrenoleukodystrophy is abbreviated as ALD or as X-ALD. The letter "X" refers to the fact that the
defective gene that causes this disorder is located on the X chromosome.
WHAT ELSE IS ADRENOLEUKODYSTROPHY KNOWN AS?
Adrenoleukodystrophy is also known as sex-linked adrenoleukodystrophy, melanodermic
leukodystrophy, and x-linked adrenoleukodystrophy.
WHAT IS THE ORIGIN OF THE TERM, ADRENOLEUKODYSTROPHY?
Adrenoleukodystrophy comes from the Latin word "ad" meaning "near," the Greek word "ren" meaning
"kidney," the Greek word "leukos" meaning "white," the Greek word "dys" meaning "difficult," and the
Greek word "trophe" meaning "nourishment." Put the words together and you get "near kidney difficult
white nourishment."